
Ein Protein aus dem Bakterium Staphylococcus aureus
Ende November gab das Google DeepMind-Team bekannt, dass sein AlphaFold-Deep-Learning-System eine beispiellose Genauigkeit bei der Lösung des Proteinfaltungsproblems erreicht hat , eines schwierigen Problems der rechnergestützten Biochemie.
Was ist das Problem und warum ist es so schwer zu lösen?
Proteine sind lange Ketten von Aminosäuren. Ihre DNA kodiert für diese Sequenzen, und RNA hilft dabei, Proteine gemäß dieser genetischen Blaupause herzustellen. Proteine werden in Form linearer Ketten synthetisiert, anschließend jedoch zu komplexen kugelförmigen Strukturen gefaltet (siehe Bild am Anfang des Artikels).
Ein Teil der Kette kann sich zu einer engen Spirale zusammenrollen. " α-Helix . "Der andere Teil kann sich hin und her biegen, um eine breite, flache Figur zu bilden," β-Faltblatt ":
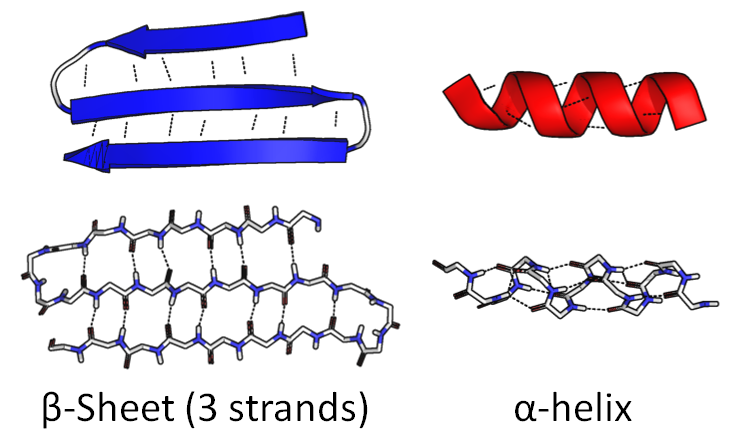
Die Aminosäuresequenz selbst wird als Primärstruktur bezeichnet . Diese Figuren werden als Sekundärstruktur bezeichnet .
Diese Komponenten selbst falten sich auch zu einzigartigen komplexen Formen. Dies wird als Tertiärstruktur bezeichnet :
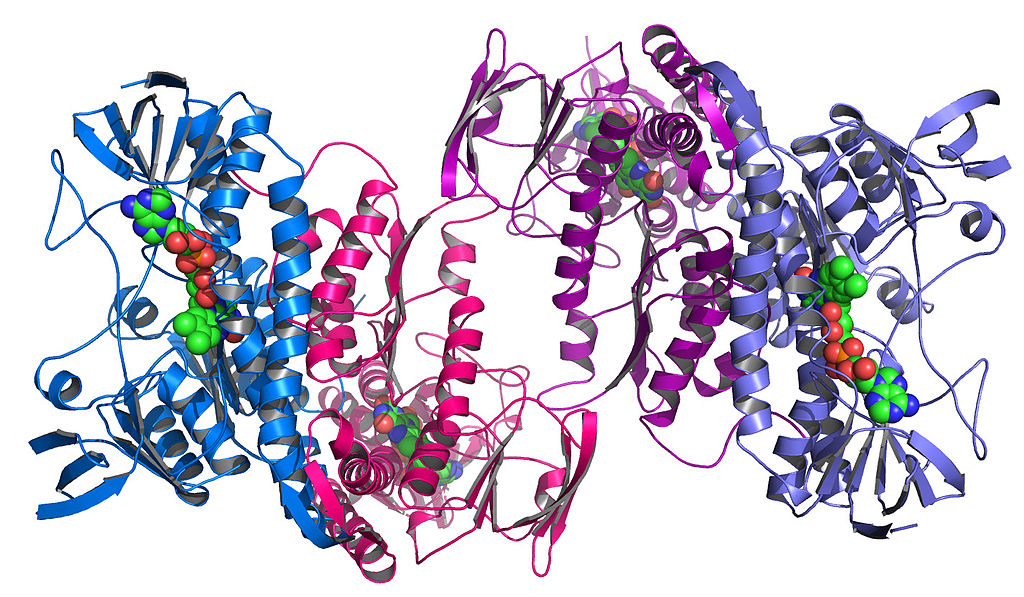
Ein Enzym, das dem RRM3-

Protein Colwellia psychrerythraea entnommen wurde.
Sieht unordentlich aus. Warum ist diese verwickelte Kugel aus Aminosäuren so wichtig?
Die Proteinstruktur ist nicht zufällig! Jedes Protein faltet sich zu einer eindeutigen, einzigartigen und weitgehend vorhersehbaren Struktur, die für eine ordnungsgemäße Funktion unerlässlich ist. Aufgrund seiner physikalischen Form ist das Protein gut für die Strukturen geeignet, an die es binden kann. Andere physikalische Eigenschaften sind ebenfalls wichtig, insbesondere die Verteilung der elektrischen Ladung über das Protein. Im Bild ist die positive Ladung blau und die negative Ladung rot dargestellt:

Oberflächenladungsverteilung auf dem Lipidträgerprotein der Pflanzen 1 von Reis
Wenn ein Protein im Wesentlichen eine selbstorganisierende Nanomaschine ist, besteht der Hauptzweck einer Aminosäuresequenz darin, ihre einzigartige Form, Ladungsverteilung und alles andere zu erzeugen, was die Funktion des Proteins bestimmt. Wie genau dieser Prozess abläuft, ist noch nicht ganz klar - heute ist es ein aktives Forschungsgebiet.
In jedem Fall ist das Verständnis der Struktur wichtig, um zu verstehen, wie sie funktioniert. Die DNA-Sequenz definiert jedoch nur die Primärstruktur des Proteins. Woher kennen wir seine Sekundär- und Tertiärstrukturen - das heißt die genaue Form, die dieses Gewirr annehmen wird?
Dieses Problem wird als Proteinfaltungsproblem bezeichnet, und es gibt zwei grundlegende Ansätze: Messung und Vorhersage.
Experimentelle Methoden können die Struktur eines Proteins messen. Dies ist jedoch nicht so einfach: Strukturen sind durch ein optisches Mikroskop nicht sichtbar. Röntgenkristallographie war lange Zeit die Hauptmethode zur Untersuchung von Strukturen. Darüber hinaus wurde die Kernspinresonanz verwendet, und kürzlich wurde eine neue Technologie eingeführt, die Kryoelektronenmikroskopie .

Röntgenbeugungsmuster der SARS-Protease
Diese Verfahren sind jedoch teuer, komplex und zeitaufwendig und funktionieren außerdem nicht mit allen Proteinen. Insbesondere falten sich in die Zellmembran eingebettete Proteine - der gleiche Angiotensin-Converting-Enzym-2 (ACE2) -Rezeptor, an den das COVID-19-Virus bindet - in die LipiddoppelschichtZellen, und es ist sehr schwierig zu kristallisieren.

Die Struktur der Zellmembran
Daher konnten wir die Struktur eines winzigen Prozentsatzes der sequenzierten Proteine zerlegen . Die universelle Proteindatenbank enthält 180 Millionen Sequenzen, während die Datenbank dreidimensionaler Proteinstrukturen nur 170.000 Positionen enthält.
Wir brauchen eine bessere Methode.
* * *
Wir erinnern daran, dass die Sekundär- und Tertiärstrukturen von Proteinen im Wesentlichen eine Funktion der Primärstruktur sind, die uns durch Sequenzierung bekannt ist. Was wäre, wenn wir die Struktur eines Proteins nicht messen, sondern vorhersagen könnten?
Dies ist die Aufgabe, die Struktur von Proteinen vorherzusagen. Computational Biochemists arbeiten seit Jahrzehnten daran.
Wie können Sie es angehen?
Der naheliegende Weg besteht darin, die Physik des Prozesses direkt zu simulieren. Wir simulieren Kräfte für jedes Atom unter Berücksichtigung seiner Position, Ladung und chemischen Bindungen. Wir zählen Beschleunigungen und Geschwindigkeiten und scrollen Schritt für Schritt durch die Entwicklung des Systems. Dies nennt man "Molekulardynamik".

Supercomputer " Anton " von DE Shaw Research

Supercomputer IBM Blue Gene
Online-Puzzle Foldit
Das Problem ist, dass dieser Ansatz äußerst rechenintensiv ist. Ein typisches Protein enthält Hunderte von Aminosäuren, dh Tausende von Atomen. Auch die Umwelt spielt eine Rolle: Beim Falten interagiert das Protein mit dem umgebenden Wasser. Daher ist es notwendig, das Verhalten von etwa 30.000 Atomen zu simulieren. In diesem Fall tritt eine elektrostatische Wechselwirkung zwischen jedem Atompaar auf, dh mit einer groben Schätzung erhalten wir 450 Millionen Paare, ein Problem mit der Komplexität O (N2). Es gibt clevere Algorithmen, die ihre Komplexität auf O (N log N) reduzieren. Zusätzlich müssen für die Simulation 10 9 -10 12 Schritte berechnet werden. Außergewöhnliche Kopfschmerzen.
Okay, aber wir müssen nicht den gesamten Faltvorgang simulieren. Ein anderer Ansatz schlägt vor, eine Struktur mit minimaler potentieller Energie zu finden. Objekte neigen normalerweise dazu, mit der geringsten Energie zur Ruhe zu kommen, daher ist dieser heuristische Ansatz gerechtfertigt. Energie kann mit demselben molekulardynamischen Modell berechnet werden, das uns die Größe der Wechselwirkungen angibt. Mit diesem Ansatz können wir eine Reihe von Kandidaten ausprobieren und die Struktur mit der geringsten Energie auswählen. Das Problem ist natürlich, woher die Strukturen stammen. Es gibt einfach zu viele von ihnen - der Molekularbiologe Cyrus Levintol hat berechnet, dass es ungefähr 10.300 geben könnten . Natürlich können Sie einen intelligenteren Ansatz als zufällige Brute Force verwenden. Aber es gibt immer noch zu viele von ihnen.
Daher wurden bereits viele Versuche unternommen, solche Berechnungen zu beschleunigen. Anton, ein Supercomputer von DE Shaw Research, verwendet spezielle Hardware - spezielle integrierte Schaltkreise. IBM verwendet auch den Blue Gene Bio-Supercomputer. Stanford startete das Folding @ Home-Projekt mit der verteilten Leistung von Heimcomputern. Das Foldit-Projekt von UW hat das Falten zu einem Spiel gemacht, um der Berechnung menschliche Intuition zu verleihen.
Lange Zeit war es jedoch keiner Technologie möglich, eine Vielzahl von Proteinstrukturen mit hoher Genauigkeit vorherzusagen. Beim zweimal jährlich stattfindenden CASP-Wettbewerb, bei dem die Ergebnisse der Algorithmen mit den experimentell gemessenen Strukturen verglichen werden, erhielten die ersten Plätze Vorhersagen mit einer Genauigkeit von 30-40%. Bis vor kurzem:

Beste Team-Median-Vorhersagegenauigkeit in der Kategorie freie Modellierung.
Wie funktioniert AlphaFold? Es verwendet mehrere tiefe neuronale Netze, um verschiedene Funktionen zu lernen, die mit jedem Protein verbunden sind. Eine der Schlüsselfunktionen besteht darin, die resultierenden Abstände zwischen Aminosäurepaaren vorherzusagen. Dies bringt den Algorithmus zur endgültigen Struktur. In einer Variante des Algorithmus (beschrieben in den Fachzeitschriften Nature and Proteins ) wurde die potenzielle Funktion dieser Vorhersage abgeleitet, auf die der einfachste Gradientenabstieg angewendet wurde, der überraschend gut funktionierte.
Der Hauptvorteil von AlphaFold gegenüber früheren Methoden besteht darin, dass keine Annahmen über Strukturen getroffen werden müssen. Einige Methoden arbeiten, indem sie Proteine in Abschnitte aufteilen, jeden einzelnen zählen und dann alles wieder zusammensetzen. AlphaFold braucht das nicht.
Anscheinend hält DeepMind das Faltproblem für gelöst, was mir als übermäßige Vereinfachung erscheint, aber auf jeden Fall sind ihre Fortschritte erheblich. Experten, die nicht mit Google verbunden sind, verwenden Beinamen wie " fantastisch " und " revolutionär ".
Die Gentechnik verfügt nun über zwei leistungsstarke Tools: CRISPR und Proteinfaltung. Vielleicht sind die 2020er Jahre für die Biotechnologie wie die 1970er Jahre für die Datenverarbeitung.
Herzlichen Glückwunsch an die DeepMind-Forscher zu diesem Durchbruch!