
In einem Papier veröffentlicht kürzlich in Physical Review Forschung zeigen wir , wie tief das Lernen , die Lösung der grundlegenden quantenmechanischen Gleichungen für reale Systeme vereinfacht. Gleichzeitig wird nicht nur ein grundlegendes wissenschaftliches Problem gelöst, sondern es eröffnen sich auch Perspektiven für die praktische Anwendung der in Zukunft erzielten Ergebnisse.
Die Forscher werden in der Lage sein, neue Materialien und Verbindungen in silico zu prototypisieren, bevor sie versuchen, sie im Labor zu synthetisieren. Habe auch den Code gepostet aus dieser Studie; Auf diese Weise können Teams für Computerphysik und Chemie auf ihrer Arbeit aufbauen und sie auf eine Vielzahl von Problemen anwenden. Im Rahmen der Studie wurde eine neue neuronale Netzwerkarchitektur, Fermionic Neural Network oder FermiNet, entwickelt, die sich gut zur Simulation des Quantenzustands großer Sammlungen von Elektronen eignet - und alle chemischen Bindungen basieren auf Elektronen. FermiNet demonstrierte zum ersten Mal, wie man mithilfe von Deep Learning die Energie von Atomen und Molekülen von Grund auf neu berechnet. Das resultierende Modell erwies sich als genau genug für die praktische Anwendung und blieb zum Zeitpunkt der Veröffentlichung des Originalartikels (Oktober 2020) die genaueste in der Branche verwendete neuronale Netzwerkmethode. Es wird davon ausgegangendass die damit verbundenen Methoden und Werkzeuge bei der Lösung grundlegender naturwissenschaftlicher Probleme hilfreich sein können. Die Autoren von FermiNet verwenden es bereits in ihrer Arbeit an die Faltung von Proteinen , die Dynamik glasartiger Verbindungen , die Quantenchromodynamik auf einem Gitter und in vielen anderen Projekten, die dazu beitragen, diese Entwicklungen in die Praxis umzusetzen.
Eine kurze Geschichte der Quantenmechanik
Wenn Sie "Quantenmechanik" erwähnen, werden Sie den Gesprächspartner wahrscheinlich wie kein anderer mit diesem Thema verwirren. Ich erinnere mich sofort an Bilder wie Schrödingers Katze, die paradoxerweise gleichzeitig lebendig und tot sein kann, sowie an Elementarteilchen, die sowohl Körperchen als auch Wellen sind. In einem Quantensystem hat ein Teilchen wie ein Elektron im Gegensatz zur klassischen Physik keinen bestimmten Ort. In der Quantenphysik wird die Position eines Elektrons durch eine Wolke von Wahrscheinlichkeiten beschrieben - das heißt, sie wird über alle Punkte verschmiert, an denen jeweils ein Elektron auftreten kann. Aufgrund dieses absurden Zustands fand Richard Feynman es möglich zu sagen: "Ich glaube, ich kann mit Sicherheit sagen, dass niemand die Quantenmechanik versteht."
Trotz all dieser unheimlichen Verrücktheit kann das Wesen der Theorie in nur wenigen sauberen Gleichungen ausgedrückt werden. Die bekannteste davon, die Schrödinger-Gleichung, beschreibt das Verhalten von Teilchen auf einer Quantenskala genauso wie Newtons Gleichungen das Verhalten von Körpern auf den bekannteren makroskopischen Skalen beschreiben. Während die Interpretation dieser Gleichung jeden dazu zwingen wird, sich den Kopf zu schnappen, ist ihre mathematische Komponente für den praktischen Gebrauch viel einfacher, wodurch das "Shut up and Count" des berühmten Professors geboren wurde, mit dem sie unangenehme philosophische Fragen von Studenten abwehrten.
Diese Gleichungen reichen aus, um das Verhalten aller uns bekannten Materie auf der Ebene von Atomen und Kernen zu beschreiben. Eine unlogische Komponente der Quantenmechanik liegt allen möglichen exotischen Phänomenen zugrunde: Supraleitung, Superfluidität, Laser und Halbleiter sind nur aufgrund von Quanteneffekten möglich. Aber selbst eine so bescheidene Sache wie eine kovalente Bindung - der Grundbestandteil aller Chemie - ist das Ergebnis von Quantenwechselwirkungen von Elektronen. Als diese Regeln in den 1920er Jahren endgültig ausgearbeitet wurden, stellten die Wissenschaftler fest, dass zum ersten Mal eine Theorie erstellt wurde, die die Arbeit der gesamten Chemie detailliert beschreibt. Im Prinzip könnten Quantengleichungen einfach für verschiedene Moleküle angepasst werden, indem die Energie des Systems berücksichtigt und dann bestimmt wird, welche Moleküle stabil sind und welche Reaktionen spontan ablaufen. Aber,Als versucht wurde, sich hinzusetzen und die Lösungen für diese Gleichungen zu berechnen, stellte sich heraus, dass dies für das einfachste Atom (Wasserstoff) und praktisch für kein anderes möglich ist. Alle anderen Berechnungen erwiesen sich als zu kompliziert.
Der schwindelerregende Optimismus jener Tage wurde von Paul Dirac wunderbar zusammengefasst:
Die grundlegenden physikalischen Gesetze, die für eine mathematische Theorie erforderlich sind, die den größten Teil der Physik und die gesamte Chemie beschreiben würde, sind bereits bekannt. Der Haken ist, dass die Anwendung dieser Gesetze in der Praxis zu komplexe Gleichungen ergibt, die wir objektiv nicht lösen können. Daher erscheint es wünschenswert, ungefähre Methoden für die praktische Anwendung der Quantenmechanik zu entwickeln.
1929
Viele nahmen Diracs Ruf auf und bald begannen Physiker, mathematische Methoden zu entwickeln, die es ermöglichen würden, das Verhalten molekularer Bindungen und anderer chemischer Phänomene auf qualitativer Ebene zu approximieren. Alles begann mit einer groben Beschreibung des Verhaltens von Elektronen - diese Informationen werden in einem Chemie-Einführungskurs untersucht. Mit dieser Beschreibung wird jedes Elektron in sein eigenes Orbital gebracht, wodurch Sie die Wahrscheinlichkeit berechnen können, dass ein Elektron an einem bestimmten Punkt in der Nähe eines Atomkerns gefunden wird. In diesem Fall hängt die Form jedes Orbitals von der durchschnittlichen Form aller anderen Orbitale ab. Da in einer solchen Beschreibung nach dem Modell des "selbstkonsistenten Feldes" angenommen wird, dass jedes Elektron nur an ein Orbital gebunden ist, vermittelt dieses Bild sehr unvollständig die realen Eigenschaften von Elektronen. Trotzdem ist es genugdie Gesamtenergie des Moleküls mit einem Fehler von nur etwa 0,5% zu bestimmen.
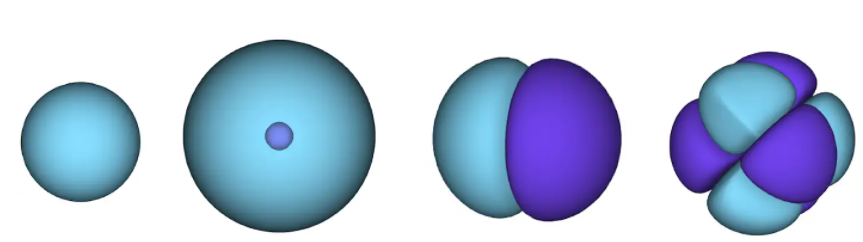
Abbildung 1 - Atomorbitale. Eine Oberfläche ist ein Bereich, in dem sich wahrscheinlich ein Elektron befindet. im blauen Bereich ist die Wellenfunktion positiv und im violetten Bereich negativ.
Leider ist für den praktizierenden Chemiker ein Fehler von 0,5% zu groß, um toleriert zu werden. Die Energie molekularer Bindungen macht nur einen kleinen Bruchteil der Gesamtenergie eines Systems aus, und die korrekte Vorhersage, ob ein Molekül stabil ist, kann häufig von nur 0,001% der Gesamtenergie des Systems oder etwa 0,2% abhängen der verbleibenden "Korrelationsenergie".
Zum Beispiel, während die Gesamtenergie der Elektronen in einem Butadienmolekül ist beträgt fast 100.000 Kilokalorien pro Mol, die Energiedifferenz zwischen den verschiedenen möglichen Konfigurationen des Moleküls beträgt nur 1 Kilokalorie pro Mol. Das heißt, wenn die natürliche Form des Butadienmoleküls korrekt vorhergesagt werden muss, ist dieselbe Genauigkeit erforderlich wie bei der Messung der Breite eines Fußballfelds mit einer Genauigkeit von einem Millimeter.
Mit der Verbreitung des elektronischen Rechnens kurz nach dem Zweiten Weltkrieg entwickelten die Wissenschaftler eine ganze Reihe von Berechnungsmethoden, die nicht als selbstkonsistente Felder bezeichnet werden konnten. Diese Methoden werden durch eine unvorstellbare Reihe von Abkürzungen gekennzeichnet, die das gesamte Alphabet abdecken. Jede dieser Methoden enthält jedoch einen Kompromiss zwischen Genauigkeit und Effizienz. Ein Extrem sind Methoden, die zwar genau sind, aber mit zunehmender Anzahl von Elektronen schlechter als exponentiell skalieren - daher sind sie nicht für die Arbeit mit den meisten, aber den kleinsten Molekülen geeignet. Im anderen Extrem gibt es Methoden, die linear skalieren, aber nicht sehr genau sind. Diese Berechnungsmethoden hatten enorme Auswirkungen auf die praktische Chemie - der Nobelpreis für Chemie von 1998 wurde an die Autoren vieler dieser Algorithmen verliehen.
Trotz der Breite der vorhandenen quantenmechanischen Berechnungswerkzeuge erforderte das Problem der effizienten Darstellung von Informationen die Entwicklung einer neuen Methode. Es ist kein Zufall, dass nur Zehntausende von Elektronen an den größten modernen quantenchemischen Berechnungen beteiligt sind (wir sprechen von den ungefährsten Methoden), während klassische Methoden chemischer Berechnungen, beispielsweise die Molekulardynamik, die Handhabung von Millionen von Elektronen ermöglichen Atome. Es ist nicht schwer, den Zustand eines klassischen Systems zu beschreiben - Sie müssen nur die Position und den Impuls jedes Teilchens verfolgen. Die Vorstellung des Zustands eines Quantensystems ist eine viel größere Herausforderung. Wir müssen jeder möglichen Konfiguration von Elektronenpositionen einen Wahrscheinlichkeitswert zuweisen. Diese Informationen werden in einer Wellenfunktion codiert.Damit können Sie jeder Elektronenkonfiguration eine positive oder negative Zahl zuweisen, und die Rechteckwellenfunktion gibt die Wahrscheinlichkeit an, mit der das System in einer solchen Konfiguration gefunden werden kann. Der Raum aller möglichen Konfigurationen ist kolossal - wenn Sie versuchen würden, ihn als Gitter mit 100 Punkten in jeder Dimension vorzustellen, wäre die Anzahl der möglichen Konfigurationen von Elektronen für ein Siliziumatom größer als die Anzahl der Atome im Universum!
Hier bieten sich tiefe neuronale Netze an. In den letzten Jahren wurden enorme Fortschritte bei der Darstellung komplexer Wahrscheinlichkeitsverteilungen mit hoher Dimensionalität unter Verwendung neuronaler Netze erzielt. Es ist jetzt bekannt, wie solche Netzwerke mit der Erwartung ihrer Skalierbarkeit effektiv trainiert werden können. Wir haben vorgeschlagen, dass diese Netzwerke, da sie bereits ihre Beweglichkeit in Trainingsfunktionen mit vielen Dimensionen bei der Lösung von Problemen aus dem Bereich der künstlichen Intelligenz bewiesen haben, möglicherweise für die Darstellung von Quantenwellenfunktionen arbeiten werden. Wir waren nicht die ersten, die solche Gedanken hatten - andere Forscher, insbesondere Giuseppe Carleo und Matthias Troyerzeigten, wie modernes Deep Learning zur Lösung idealisierter Quantenprobleme anwendbar ist. Wir wollten neuronale Netze verwenden, um realistischere Probleme in der Chemie und Festkörperphysik anzugehen, was bedeutete, dass wir Elektronen in unseren Berechnungen berücksichtigen mussten.
Bei der Arbeit mit Elektronen gibt es nur eine Einschränkung. Elektronen müssen dem Pauli-Ausschlussprinzip folgen, dh zwei Elektronen können nicht gleichzeitig am selben Ort sein. Tatsache ist, dass Elektronen Elementarteilchen aus den Fermionen sind, aus denen der Großteil der ersten Materiesteine besteht, insbesondere Protonen, Neutronen, Quarks, Neutrinos usw. Ihre Wellenfunktion muss antisymmetrisch sein - wenn Sie zwei Elektronen austauschen, wird die Wellenfunktion mit -1 multipliziert. Somit besteht eine Wahrscheinlichkeit von Null, dass zwei Elektronen übereinander sitzen, da die Wahrscheinlichkeit dafür (und die entsprechende Wellenfunktion) gleich Null ist.
Daher war es notwendig, ein neuronales Netzwerk eines neuen Typs zu entwickeln, das in Bezug auf die Eingabe, die in es eingeht, antisymmetrisch wäre. Wir haben es Fermionic Neural Network oder FermiNet genannt. Bei den meisten quantenchemischen Methoden wird die Antisymmetrie unter Verwendung einer als Determinante bezeichneten Funktion eingeführt. Die Determinante ist eine Matrix mit der folgenden Eigenschaft: Wenn Sie zwei ihrer Zeilen vertauschen, wird die Ausgabe genau wie die Wellenfunktion von Fermionen mit -1 multipliziert. Sie können eine Reihe von Einelektronenfunktionen verwenden, diese für jedes Elektron in Ihrem System berechnen und dann alle Ergebnisse in eine Matrix einpassen. In diesem Fall ist die Determinante der Matrix eine wirklich antisymmetrische Wellenfunktion. Die Hauptbeschränkung dieses Ansatzes besteht darin, dass die resultierende Funktion - Slater Determinant genannt - nicht allgemein anwendbar ist.Die Wellenfunktionen realer Systeme sind normalerweise viel komplexer. Typischerweise werden große lineare Kombinationen von Slater-Determinanten - manchmal Millionen oder mehr - verwendet, um dieses Problem zu beheben, und dann werden einige einfache Korrekturen basierend auf Elektronenpaaren vorgenommen. Selbst dann ist das System möglicherweise nicht genau genug, um Energien zu berechnen.

2 – . – , 1. 1 2 , , -1. .
Tiefe neuronale Netze sind linearen Kombinationen von Basisfunktionen bei der Darstellung komplexer Funktionen in ihrer Effizienz oft weit überlegen. In FermiNet wird diese Überlegenheit erreicht, indem jede der Funktionen in die Determinante, die Funktion aller Elektronen, eingeführt wird. Diese Methode ist viel leistungsfähiger als die Verwendung von Ein- und Zwei-Elektronen-Funktionen. FermiNet bietet für jedes Elektron einen eigenen Informationsstrom. Ohne Berücksichtigung von Wechselwirkungen zwischen diesen Flüssen wäre das Netzwerk nicht aussagekräftiger als die übliche Slater-Determinante. Um mehr zu erreichen, mitteln wir die von allen Streams auf jeder der Netzwerkschichten gesammelten Informationen und geben diese Informationen an jeden der Streams an die nächste Schicht weiter. Dementsprechend haben solche Strömungen geeignete Symmetrieeigenschaften, um eine antisymmetrische Funktion zu erzeugen.
Informationen zu jeder der Schichten in grafischen neuronalen Netzen werden auf ähnliche Weise aggregiert . Im Gegensatz zu Slaters Determinanten sind FermiNet-Netzwerke universelle Funktionsapproximatoren , zumindest solange die Schichten neuronaler Netzwerke breit genug bleiben. Das heißt, wenn wir diese Netzwerke richtig trainieren können, können sie eine fast exakte Lösung für die Schrödinger-Gleichung ergeben.
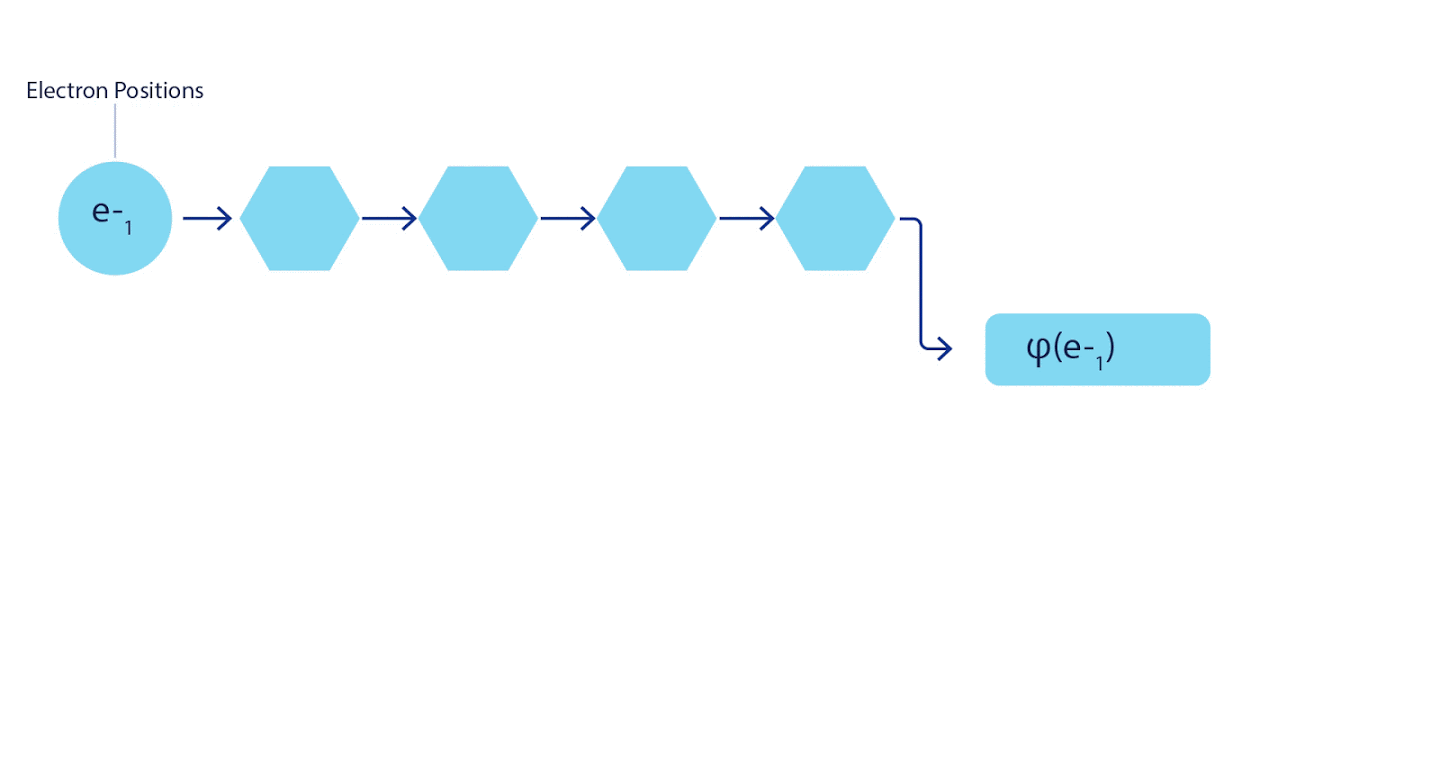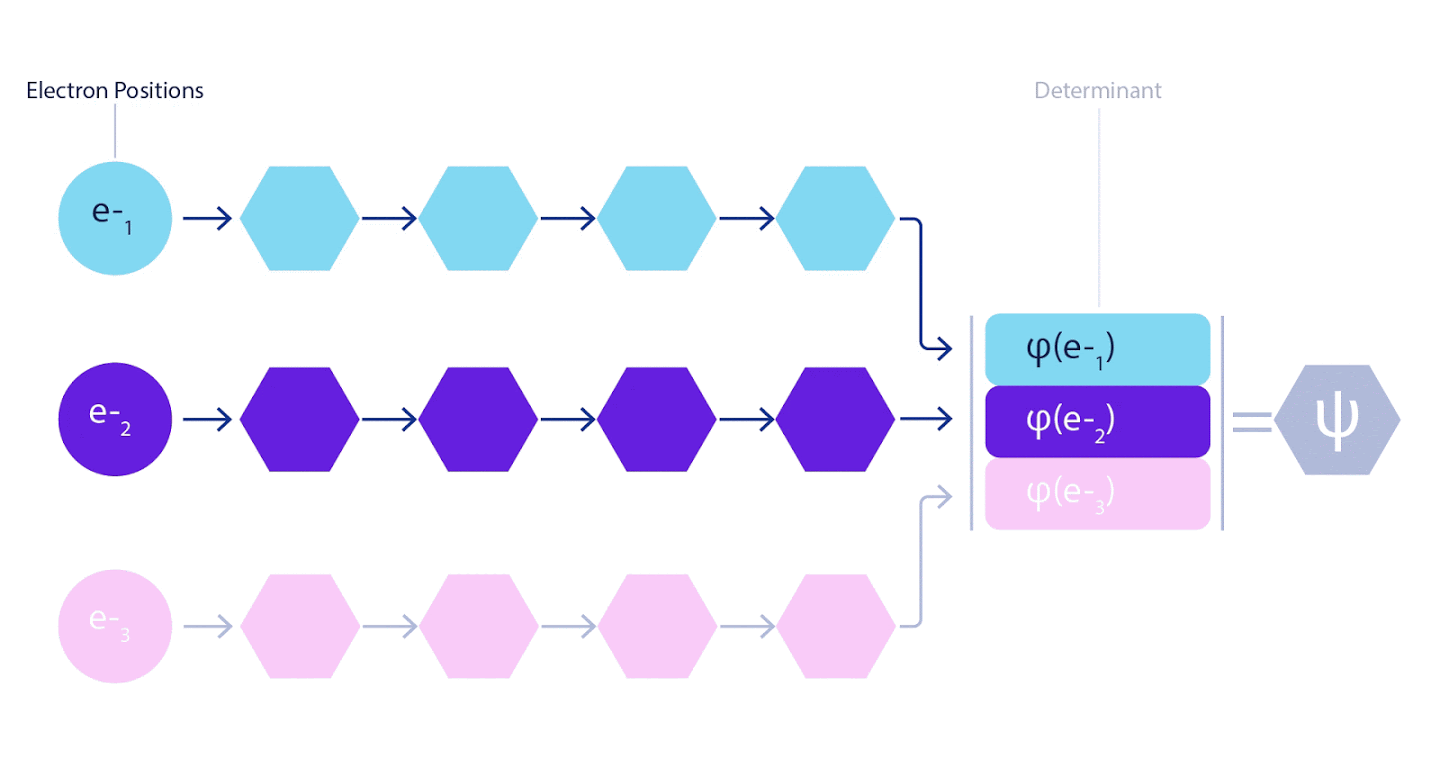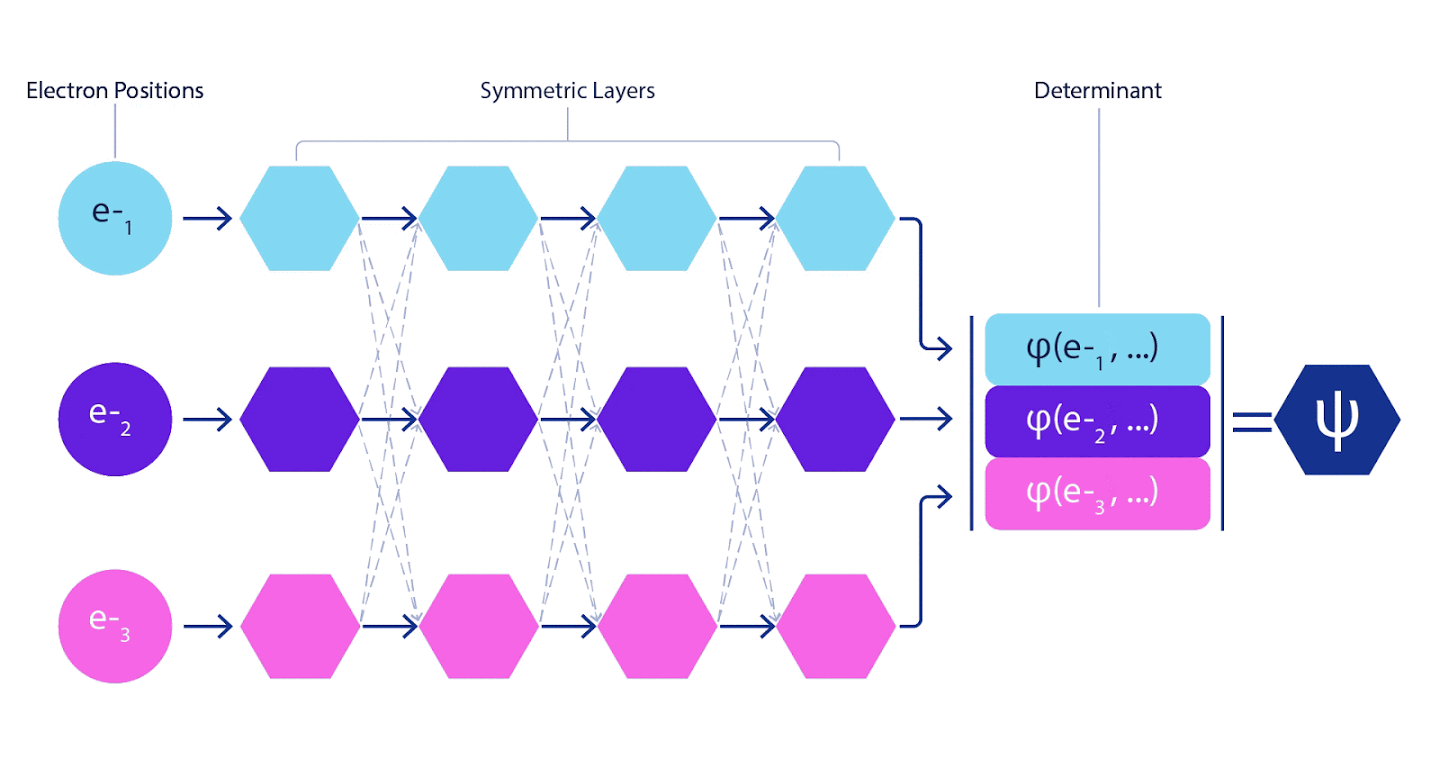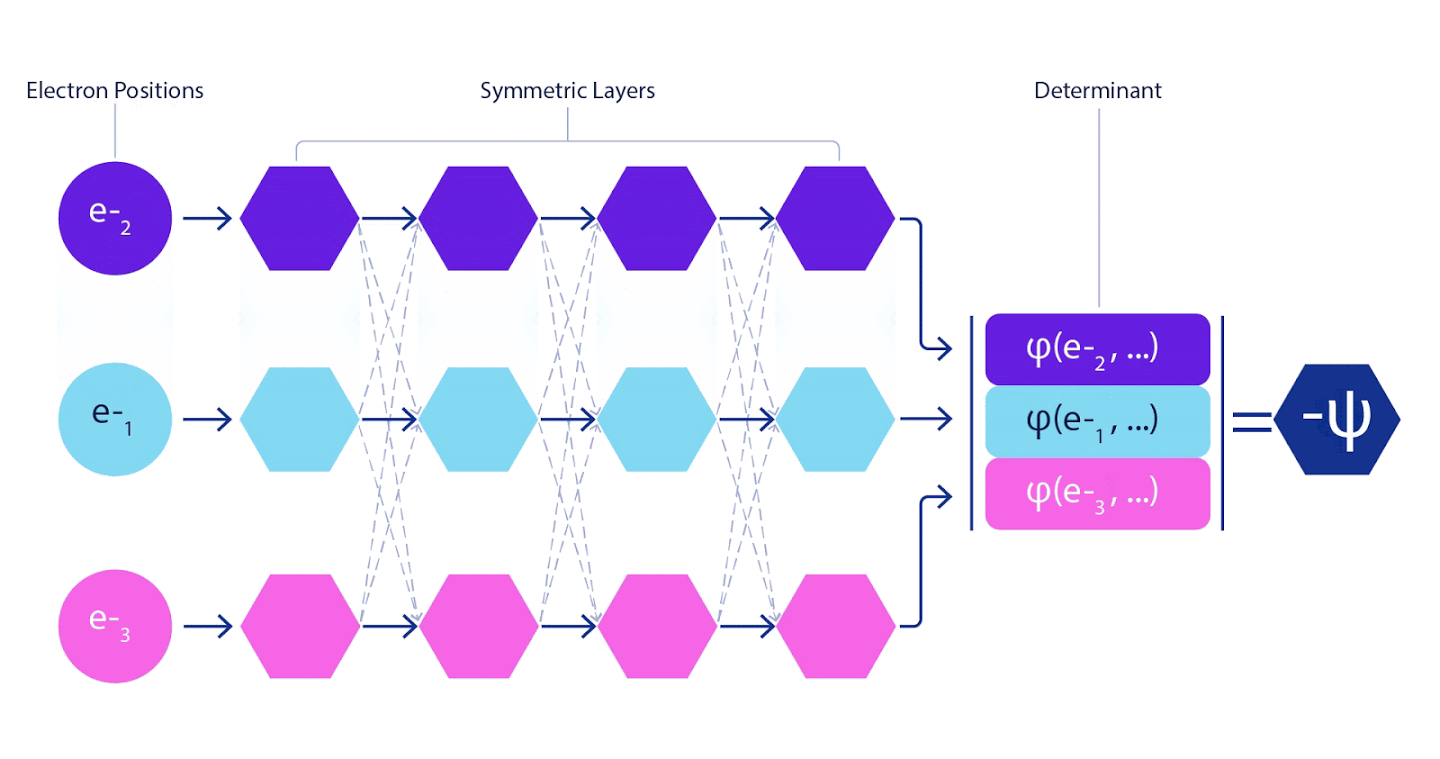
3 – FermiNet. (, ) . FermiNet , , , . , - -1.
Wir passen das FermiNet-Netzwerk an und minimieren so die Systemenergie. Um dies genau zu tun, müssten wir die Wellenfunktion in allen möglichen Elektronenkonfigurationen berechnen, also müssten wir dies ungefähr tun. Daher nehmen wir eine zufällige Stichprobe von Elektronenkonfigurationen, berechnen die Energie lokal für jede Variante der Elektronenordnung und minimieren diese Energie, nicht die wahre. Diese Methode wird "Monte Carlo" genannt, weil sie den Aktionen eines Casinospielers ähnelt, der immer wieder würfelt. Da die Rechteckwellenfunktion es ermöglicht, eine bestimmte Konfiguration von Partikeln an jedem Ort zu beobachten, ist es am bequemsten, Proben der Wellenfunktion selbst zu erzeugen - im Wesentlichen um den Vorgang der Beobachtung von Partikeln zu simulieren.
Während die meisten neuronalen Netze auf einige externe Daten trainiert werden, generiert in unserem Fall das neuronale Netz selbst die Eingabe, die für das Training in das Netzwerk eingeht. Die Situation ist ein bisschen so, als würde man sich an den Haaren aus einem Moor ziehen und bedeutet, dass wir keine anderen Trainingsdaten benötigen als die Positionen der Atomkerne, um die Elektronen tanzen. Die Grundidee, bekannt als Variational Quantum Monte Carlo Method (kurz VMC), gibt es in der Wissenschaft seit den 1960er Jahren und wird allgemein als billige, aber nicht sehr genaue Methode zur Berechnung der Energie eines Systems angesehen. Durch Ersetzen einfacher Wellenfunktionen basierend auf Slater-Determinanten durch Funktionen von FermiNet ist es uns gelungen, die Genauigkeit dieses Ansatzes auf allen von uns betrachteten Systemen radikal zu verbessern.
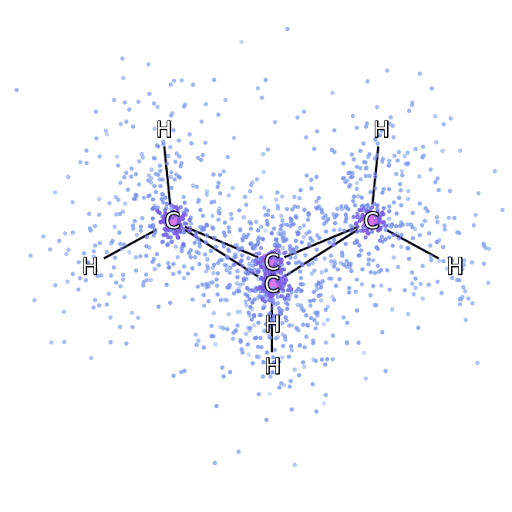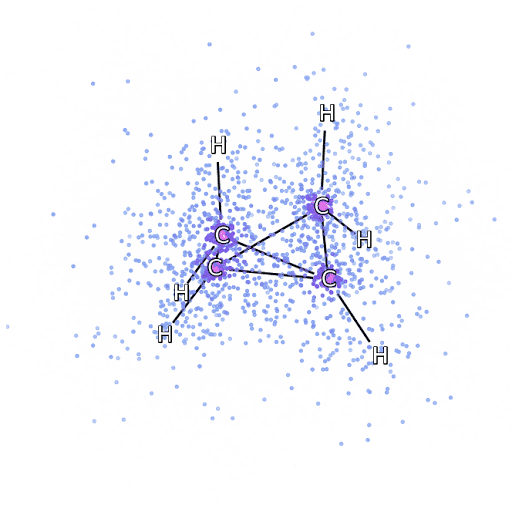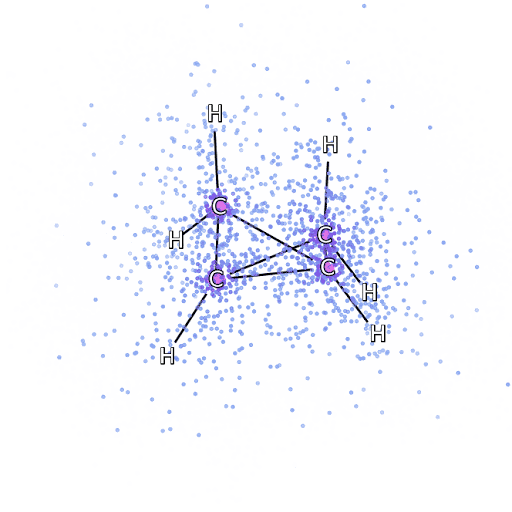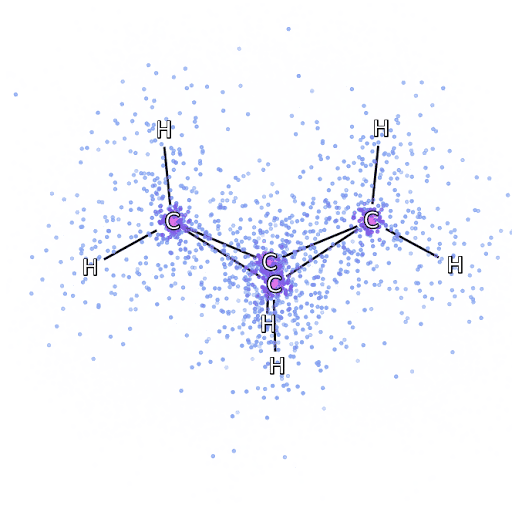
4 – , FermiNet, .
Um sicherzustellen, dass FermiNet wirklich ein Durchbruch in seinem Fachgebiet ist, haben wir zunächst einfache, gut untersuchte Systeme untersucht, beispielsweise Atome aus der ersten Zeile des Periodensystems (von Wasserstoff bis Neon). Dies sind kleine Systeme - 10 Elektronen oder weniger -, daher eignen sie sich für die Forschung mit den genauesten (aber exponentiell komplexeren) Methoden. FermiNet übertrifft vergleichbare VMC-Berechnungen bei weitem und kann Fehler im Vergleich zu exponentiell skalierbaren Berechnungen häufig halbieren oder mehr. In größeren Systemen werden Methoden, die exponentiell komplexer werden, nicht mehr anwendbar, daher haben wir die gekoppelte Cluster-Methode als Referenz verwendet. Diese Methode funktioniert gut bei Molekülen mit stabilen Konfigurationen, rutscht jedoch ab, wenn die Bindungen gedehnt oder beschädigt werden.und solche Faktoren sind entscheidend für das Verständnis chemischer Reaktionen. Die in dieser Studie verwendete Methode des verbundenen Clusters funktioniert zwar viel besser als exponentiell, funktioniert jedoch mit mittelgroßen Molekülen immer noch so gut wie möglich. Wir haben FermiNet auf immer größere Moleküle angewendet, von Lithiumhydrid bis Bicyclobutan - es war mit 30 Elektronen das größte System, das wir uns angesehen haben. Bei den kleinsten Molekülen konnte FermiNet einen erstaunlichen Unterschied von 99,8% zwischen der Energie gebundener Cluster und der Energie einer einzelnen Slater-Determinante feststellen. Im Fall von Bicyclobutan hat FermiNet immer noch 97% oder mehr dieser Korrelationsenergie erfasst - eine enorme Leistung für einen angeblich "billigen, aber ungenauen" Ansatz.Die in dieser Studie verwendete Methode des verbundenen Clusters funktioniert zwar viel besser als exponentiell, funktioniert jedoch mit mittelgroßen Molekülen immer noch so gut wie möglich. Wir haben FermiNet auf immer größere Moleküle angewendet, von Lithiumhydrid bis Bicyclobutan - es war mit 30 Elektronen das größte System, das wir uns angesehen haben. Bei den kleinsten Molekülen konnte FermiNet einen erstaunlichen Unterschied von 99,8% zwischen der Energie gebundener Cluster und der Energie einer einzelnen Slater-Determinante feststellen. Im Fall von Bicyclobutan hat FermiNet immer noch 97% oder mehr dieser Korrelationsenergie erfasst - eine enorme Leistung für einen angeblich "billigen, aber ungenauen" Ansatz.Die in dieser Studie verwendete Methode des verbundenen Clusters funktioniert zwar viel besser als exponentiell, funktioniert jedoch mit mittelgroßen Molekülen immer noch so gut wie möglich. Wir haben FermiNet auf immer größere Moleküle angewendet, von Lithiumhydrid bis Bicyclobutan - es war mit 30 Elektronen das größte System, das wir uns angesehen haben. Bei den kleinsten Molekülen konnte FermiNet einen erstaunlichen Unterschied von 99,8% zwischen der Energie gebundener Cluster und der Energie einer einzelnen Slater-Determinante feststellen. Im Fall von Bicyclobutan hat FermiNet immer noch 97% oder mehr dieser Korrelationsenergie erfasst - eine enorme Leistung für einen angeblich "billigen, aber ungenauen" Ansatz.In der beschriebenen Studie angewendet, arbeitet jeder gleichermaßen maximal mit mittelgroßen Molekülen. Wir haben FermiNet auf immer größere Moleküle angewendet, von Lithiumhydrid bis Bicyclobutan - es war mit 30 Elektronen das größte System, das wir uns angesehen haben. Bei den kleinsten Molekülen konnte FermiNet einen erstaunlichen Unterschied von 99,8% zwischen der Energie gebundener Cluster und der Energie einer einzelnen Slater-Determinante feststellen. Im Fall von Bicyclobutan hat FermiNet immer noch 97% oder mehr dieser Korrelationsenergie erfasst - eine enorme Leistung für einen angeblich "billigen, aber ungenauen" Ansatz.In der beschriebenen Studie angewendet, arbeitet jeder gleichermaßen maximal mit mittelgroßen Molekülen. Wir haben FermiNet auf immer größere Moleküle angewendet, von Lithiumhydrid bis Bicyclobutan - es war mit 30 Elektronen das größte System, das wir uns angesehen haben. Bei den kleinsten Molekülen konnte FermiNet einen erstaunlichen Unterschied von 99,8% zwischen der Energie gebundener Cluster und der Energie einer einzelnen Slater-Determinante feststellen. Im Fall von Bicyclobutan hat FermiNet immer noch 97% oder mehr dieser Korrelationsenergie erfasst - eine enorme Leistung für einen angeblich "billigen, aber ungenauen" Ansatz.von Lithiumhydrid bis Bicyclobutan - es war das größte System, das wir in Betracht gezogen haben, es hat 30 Elektronen. Bei den kleinsten Molekülen konnte FermiNet einen erstaunlichen Unterschied von 99,8% zwischen der Energie gebundener Cluster und der Energie einer einzelnen Slater-Determinante feststellen. Im Fall von Bicyclobutan hat FermiNet immer noch 97% oder mehr dieser Korrelationsenergie erfasst - eine enorme Leistung für einen angeblich "billigen, aber ungenauen" Ansatz.von Lithiumhydrid bis Bicyclobutan - es war das größte System, das wir in Betracht gezogen haben, es hat 30 Elektronen. Bei den kleinsten Molekülen konnte FermiNet einen erstaunlichen Unterschied von 99,8% zwischen der Energie gebundener Cluster und der Energie einer einzelnen Slater-Determinante feststellen. Im Fall von Bicyclobutan hat FermiNet immer noch 97% oder mehr dieser Korrelationsenergie erfasst - eine enorme Leistung für einen angeblich "billigen, aber ungenauen" Ansatz.aber ungenauer "Ansatz.aber ungenauer "Ansatz.

Abbildung 5 ist eine grafische Darstellung des Anteils der Korrelationsenergie, die FermiNet bei der Arbeit mit Molekülen korrekt erfasst. Der violette Balken markiert 99% Korrelationsenergie. Von links nach rechts: Lithiumhydrid, Stickstoff, Ethylen, Ozon, Ethanol und Bicyclobutan.
Während gekoppelte Cluster-Methoden mit stabilen Molekülen gut funktionieren, hat die eigentliche "Schneide" der Computerchemie damit zu tun, zu verstehen, wie sich Moleküle dehnen, verdrehen und brechen. Bei der Lösung solcher Probleme schlagen verbundene Clustermethoden häufig fehl. Daher müssen Sie das Ergebnis mit möglichst vielen Kontrollbeispielen vergleichen, um sicherzustellen, dass die Antwort konsistent ist. Im Rahmen des beschriebenen Experiments wurden zwei gestreckte Kontrollsysteme betrachtet - ein Stickstoffmolekül (N 2)) und eine Wasserstoffkette von 10 Atomen (H 10 ). Im Stickstoffmolekül ist die Bindung besonders komplex, da 3 Elektronen von jedem Atom daran beteiligt sind.
Die Wasserstoffkette wiederum ist interessant, um zu verstehen, welche Eigenschaften Elektronen in Materialien aufweisen , um beispielsweise vorherzusagen, ob ein bestimmtes Material Elektrizität leitet oder nicht. In beiden Systemen funktionierte die Methode der verbundenen Cluster im Gleichgewicht gut, stieß jedoch beim Strecken der Bindungen auf Schwierigkeiten. Herkömmliche VMC-Methoden zeigten nicht in allen Beispielen eine gute Leistung. FermiNet erwies sich jedoch unabhängig von der Verbindungslänge als eine der besten Methoden aller untersuchten Methoden.
Fazit
Wir glauben, dass FermiNet der Beginn großer Fortschritte bei der Synthese von Deep-Learning-Methoden und rechnergestützter Quantenchemie ist. Die meisten Systeme, mit denen FermiNet bisher überprüft wurde, sind gut verstanden und verstanden. Aber genau wie die ersten guten Ergebnisse mit Deep Learning in anderen Bereichen zu einem Anstieg der Forschung und zu raschen Fortschritten geführt haben, wird dies hoffentlich auch bei FermiNet der Fall sein und es werden Ideen für neue, noch bessere neuronale Netzwerkarchitekturen entstehen. Bereits nachdem die beschriebene Arbeit auf arXiv gepostet wurde, wurden andere Gruppenteilten ihre Ansätze zur Anwendung von Deep Learning zur Lösung von Problemen, an denen viele Elektronen beteiligt sind. Darüber hinaus haben wir uns gerade erst mit der rechnergestützten Quantenphysik befasst und planen, mit FermiNet komplexe Probleme auf dem Gebiet der Materialwissenschaften und der Festkörperphysik zu lösen.
Der wissenschaftliche Artikel ist hier und der Code kann hier eingesehen werden . Die Autoren möchten Jim Kinwin, Adam Kine und Dominic Barlow für ihre Hilfe bei der Erstellung der Zeichnungen danken.